5.2 Task setting
5.2.1 Retrieve public samples for re-analysis
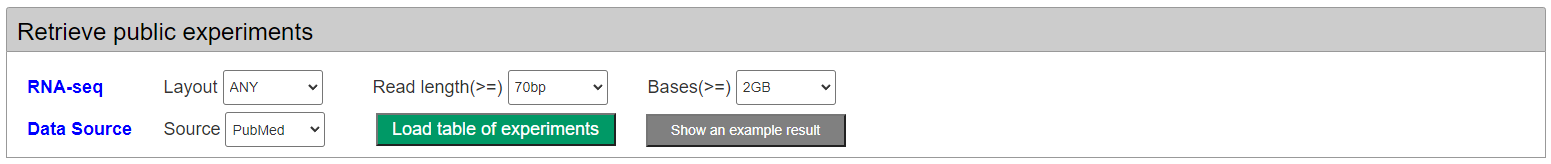
Firstly, users can retrieve the public RNA-seq samples from the platform database by multiple conditions, such as RNA-seq read length, layout, volumen and datasource.
5.2.2 Set groups
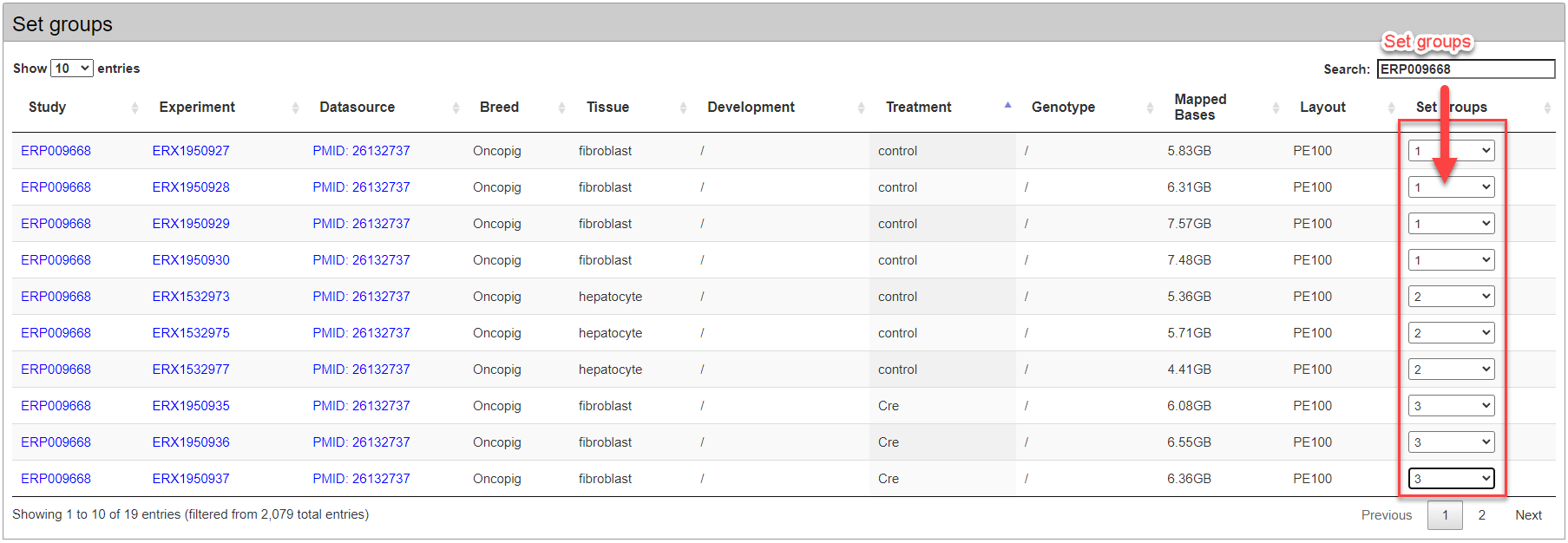
In the retrieved public samples, users should set multiple experimental groups by cultivar, genotype, tissue, development and treatment.
5.2.3 upload samples for co-analysis
To perform co-analysis with public RNA-seq samples, users can upload their RNA-seq data through the following interface. Refer to the
"co-analysis" page to see how to prepare user's own RNA-seq data.
5.2.4 Set group traits
Users can set trait discription to each group. If without trait setting, the group traits will be identified by character such as A, B, C, D, ...
5.2.5 set methods and parameters
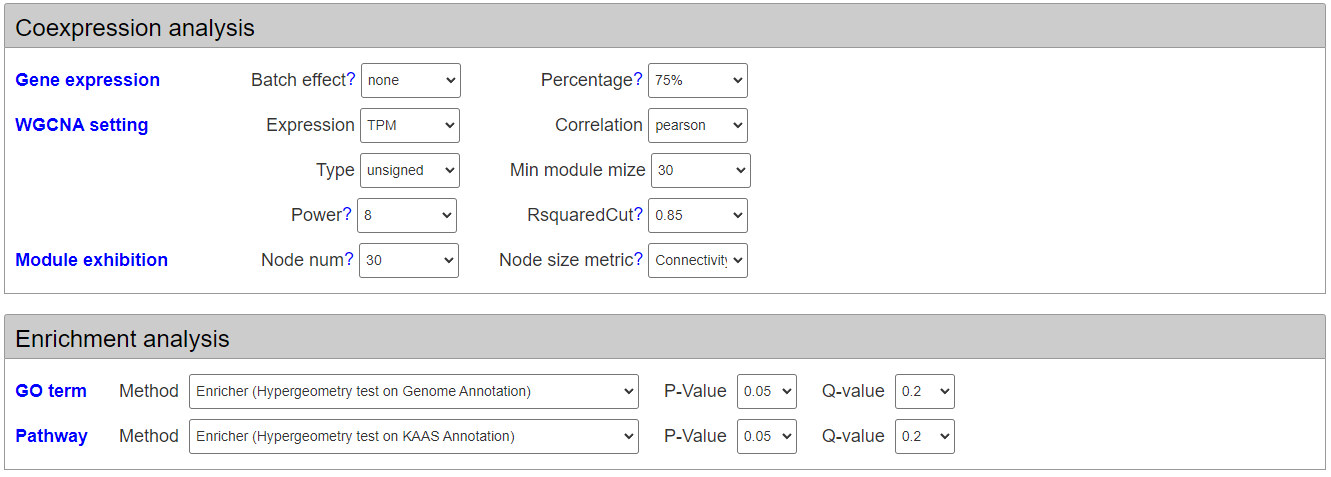
To run WGCNA, users should set some parameters. For example, whether to remove batch effect from different studies and which type of gene expression levels is used to construct co-expression network. Hover the mouse over relative parameter to learn the function and detail of corresponding parameter.
Moreover, users can select different methods for enrichment analysis on differentially expressed genes and differentially spliced genes. The enrichment analysis methods include Hyergeometry test and Gene Set Enrichment Analysis. The subject of enrichment analysis include gene ontology and KEGG pathways.
5.2.6 job request
The analysis task must be manually confirmed by email to avoid tasks from non-human submission. Users must offer email address to accept confirmation notice or retrieve analysis results.